裁切序列是 NGS 品質控管重要的一環,諸如建庫時添加的人工序列和序列末端品質低落的片段等都是要移除的目標。Trimmomatic 是專門用於 Illumina 定序資料的品管軟體,它可以發揮以下功能:
從 3' 端切除建庫時添加的人工序列
依照四種不同的模式裁切品質不佳的序列
依照平均定序品質分數篩選序列
一、切除人工序列
Trimmomatic 移除技術序列的有簡單模式和回文模式,兩種都是以用戶提供的技術序列清單,比對研究資料。一旦比對得分超過設定的閾值,便會切除之。
1. 簡單模式
簡言之,Trimmomatic 的演算法會將用戶提供的人工序列清單,沿著樣本序列的 5' 端掃描到 3' 端,計算參考序列與對應區域的匹配分數。一旦匹配分數高於設定的標準,便切除比對處與其後的片段。
雖然原則上只需切除 illumina 序列 3' 的人工序列。(參考:Adapter trimming: Why are adapter sequences trimmed from only the 3' ends of reads?),但若發生讀長大於增幅片段長度的情形,便會出現 5' 的人工序列。
此時,簡單模式的裁切方法可能會棄掉整條序列(因為一開始比對到人工序列),所以得靠迴文模式來解決。

2. 迴文模式 (palindrome mode)
迴文模式則利用雙端定序的結果會彼此重合的性質,移除因讀穿待測序列而觸及的人工序列。簡言之,順向股與逆向股互相比對,一旦發現兩端形成迴文結構,便將之移除

二、切除低品質的片段
- 不管定序品質與否,一律從特定位點切除其餘序列
- 由特定端起,切除定序品質低於標準位點以後的序列
- 由特定端起,切除定序品質低於標準的區段以後的序列

三、關於比對方式的疑問
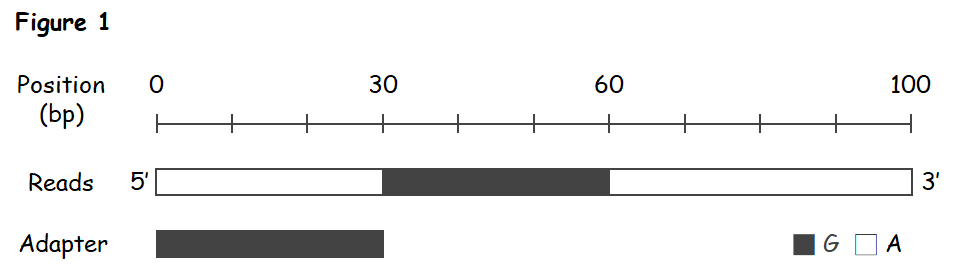
模擬序列長 100 bp,在 30 - 60 bp 的位置是長 30bp 的轉接子,圖中白色代表 A,黑色代表 G,每個鹼基的品質分數都是 30 分。
為了切除模擬資料中長度 30 bp 的轉接子,裁切標準至多設定為 30 × log 4 = 18.1。在這種條件下,只有完全匹配的片段才會自序列切除。
模擬資料的品質分數是 30 分,所以錯誤配對將扣 3 分。換句話說,要有 3 / 0.6 = 5 個正確匹配的鹼基才能抵一個錯誤配對的鹼基扣掉的分屬。所以我預期在模擬資料中,即使標準設定為 0 也不至於切錯地方。
然而實際測試結果卻不如預期 (Table 1)。當把 simple clip threshold 設定為 0 時,Trimmomatic 的裁切處是 16 bp 位置,但我估計的配對分數是 16 × log 4 - 14 × 3 = - 32.4,應該是不會切除才對。
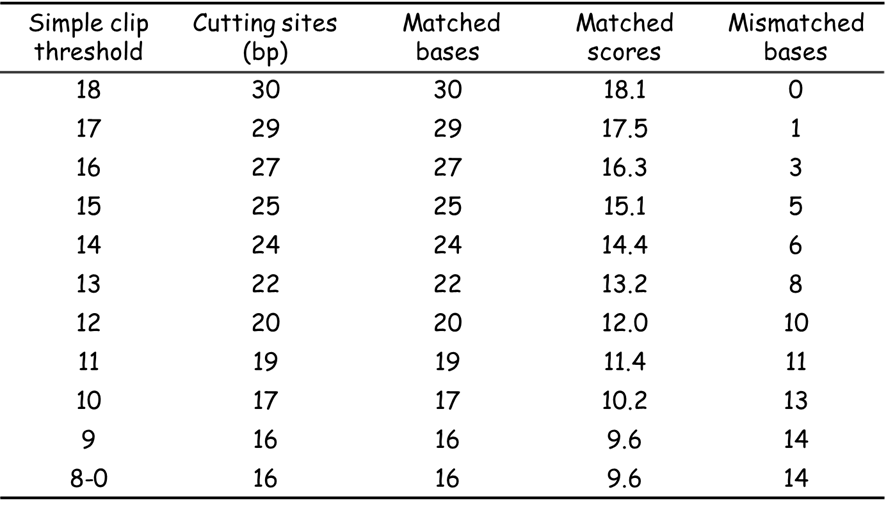
重新閱讀 Trimmomatic 的論文還是不了解其原因,所以嘗試設定不同的 simple clip threshold 檢視裁切的產物。結果如 Table 1,如果 threshold 低於上限 (18.1) 皆提早切除了部分正常序列。
這個結果看起來,Trimmomatic 在計算分數時似乎沒有扣分,只要配對達到標準就切掉了。由於沒有看原始碼,所以我也不清楚裁切的準則為何,但這種情況表示 Trimmomatic 的結果不容易讓我理解,以至於我無法預測裁切的結果如何。
除了這個問題,為提升運算速度 Trimmomatic 採兩階段式篩選 。我原先以為 seed mismatch 和結果無關,不過調整了 seed mismatch 後發現,當 threshold 設定較低的時候,mismatch 才會影響切割的結果 ,mismatch 設越高,需要越多配對。
是以,這種方式會影響裁切結果(難以預測的方式),即使樣本中出現完全一致的序列,Trimmomatic 也不會剛好切在那個地方。
(我使用的是 trimmomatic-0.39 ,參數為 SE -phred33 ILLUMINACLIP:<fastaWithAdaptersEtc>:2:30:<simple clip threshold> ,腳本與模擬資料的 GOOGLE 雲端連結為:https://tinyurl.com/uekzncy)
註
[1] Trimmomatic: a flexible trimmer for Illumina sequence data (URL: https://academic.oup.com/bioinformatics/…/30/15/2114/2390096)
[2] "If the seeds are within the user-specified distance, the full alignment scoring algorithm is used. Matching bases are scored as forumla , which is ∼0.602, while mismatches are penalized depending on their quality score, by forumla , which can thus vary from 0 to 4. This results in a higher penalty for bases that are believed to be highly accurate. ‘Simple’ mode aligns each read against each technical sequence, using local alignment. This is implemented by finding the highest scoring region within the alignment, and thus may omit divergent regions on the ends."
沒有留言:
張貼留言